
Forskere ved Stanford University avdekker en ny mekanisme som kan forklare hukommelsestap ved Alzheimers sykdom. I stedet for å være passive ofre, aktiverer nevroner selv en sletteprosess som fjerner synapser. Funnene peker mot nye mål for behandling.
Nevroner sletter egne forbindelser
Et team ledet av professor Carla Shatz viser at hjerneceller aktivt kan fjerne synapser som svar på sykdomssignaler. Arbeidet utfordrer den dominerende antagelsen om at hjernens immunceller (mikroglia) alene styrer synapsetapet ved Alzheimer.
«Nevroner er ikke uskyldige tilskuere. De er aktive deltakere.»
Samme bryter for amyloid og betennelse
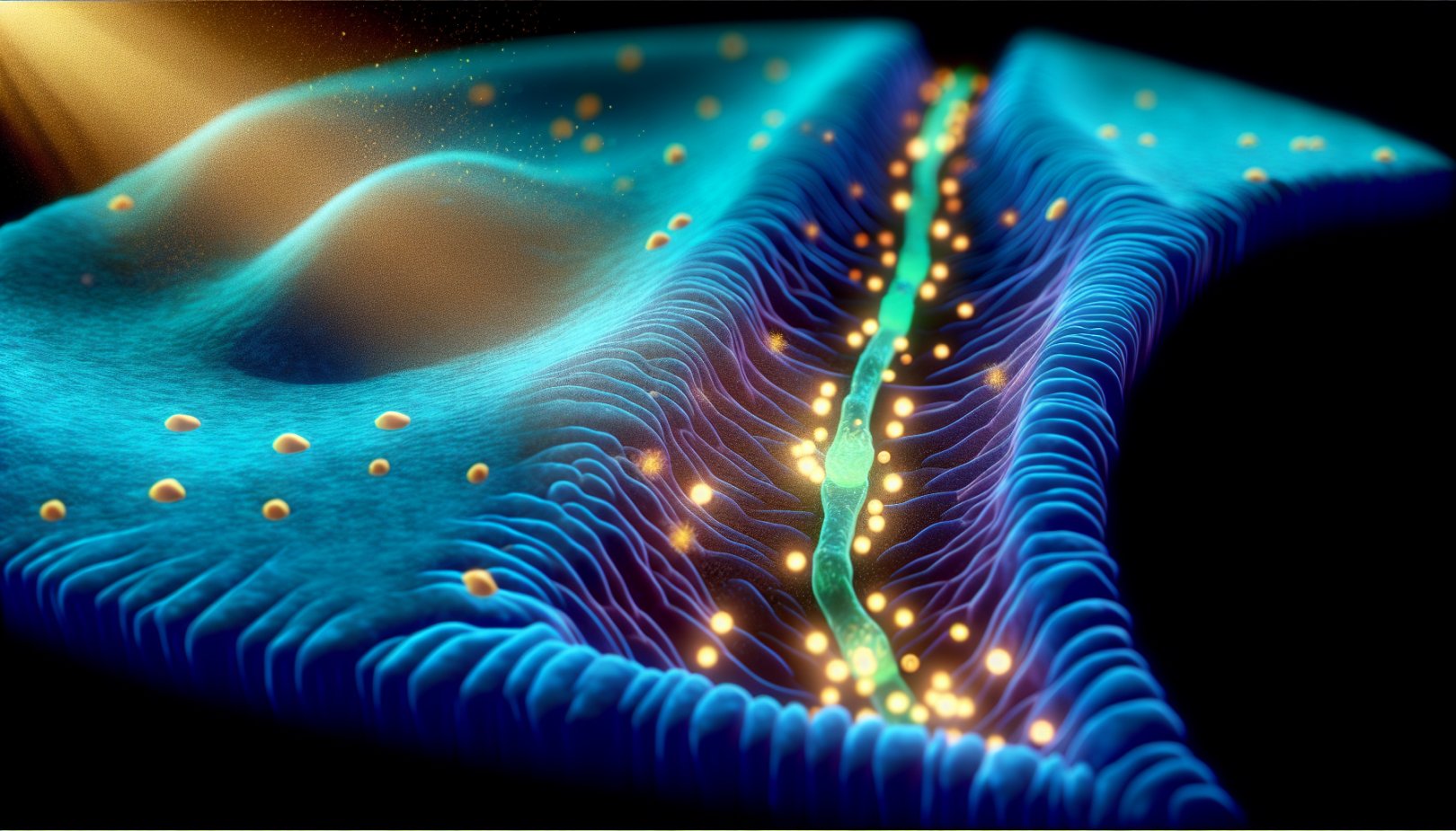
Studien identifiserer reseptoren LilrB2 som en slags bryter i nevronene. Både amyloid beta-proteiner og betennelse i hjernen ser ut til å virke gjennom denne samme mekanismen: Når LilrB2 aktiveres, får nevronene beskjed om å fjerne synapser — kontaktpunktene som er avgjørende for hukommelse og læring.
C4d: Fra «uvirksomt» fragment til nøkkelspiller
Det mest oppsiktsvekkende funnet er molekylet C4d, et spalteprodukt fra immunsystemets komplementkaskade, tidligere antatt å være uten funksjon. Studien viser at C4d binder seg til LilrB2 med svært høy affinitet og utløser synapsefjerning.
- Når forskere injiserte C4d i hjernen til friske mus, ble synapser systematisk fjernet fra nevronene.
- Hos mennesker øker C4d og C4 med alderen, og enda mer ved Alzheimers sykdom.
- I hjernecortex hos mennesker finnes C4d og LilrB2 sammen ved eksitatoriske synapser — og også sammen med amyloid beta i Alzheimer-hjerner.
«Dette var helt overraskende for et molekyl som forskere tidligere trodde ikke hadde noen funksjon i det hele tatt», sier Shatz.
- Synapser: Kontaktpunkter der hjerneceller kommuniserer; grunnlaget for hukommelse og læring.
- LilrB2: Reseptor på nevroner som kan utløse fjerning av synapser.
- C4/C4d: Deler av immunsystemets komplementkaskade; C4d er et spalteprodukt som her viser seg å være aktivt.
- Amyloid beta: Protein som danner plakk og er sentralt i Alzheimers sykdom.
Utfordrer rådende forklaring
Funnene flytter oppmerksomheten fra mikroglia som hovedaktører til selve nevronene. Dataene tyder på at nevroner direkte kontrollerer synapsetap via LilrB2 når de eksponeres for amyloid beta og betennelsessignaler.
Hva betyr dette for behandling?
I dag retter de FDA-godkjente Alzheimer-behandlingene seg mot å bryte ned amyloid-plakk i hjernen. Effekten er begrenset, og bivirkningene kan være betydelige, som hodepine og hjerneblødninger. Et mer lovende spor kan være å målrette reseptorer som LilrB2 for å beskytte synapsene — og slik bevare selve hukommelsen.
- Publisert i Proceedings of the National Academy of Sciences (PNAS).
- Delvis støttet av Knight Initiative for Brain Resilience ved Wu Tsai Neurosciences Institute ved Stanford.
Oppdagelsen fra Shatz og kolleger peker på en presis, nevronstyrt mekanisme for synapsetap i Alzheimer — via LilrB2 og C4d. Neste steg blir å teste om målrettet blokkering av denne bryteren kan stoppe tapet av synapser og bevare kognitiv funksjon. Kilder: ScienceDaily, Wu Tsai Neurosciences Institute Stanford University, Proceedings of the National Academy of Sciences (PNAS), Stanford Bio-X.





Kommentarer
0 kommentarer
Vi godtar kun kommentarer fra registrerte brukere. Dette gjør vi for å opprettholde en trygg og respektfull debatt, samt for å unngå spam og misbruk. Registrering er gratis og tar bare noen sekunder.
Du må være innlogget for å kommentere. Logg inn eller registrer deg for å delta i diskusjonen.